Janggu - Deep learning for Genomics¶





Janggu is a python package that facilitates deep learning in the context of genomics. The package is freely available under a GPL-3.0 license.
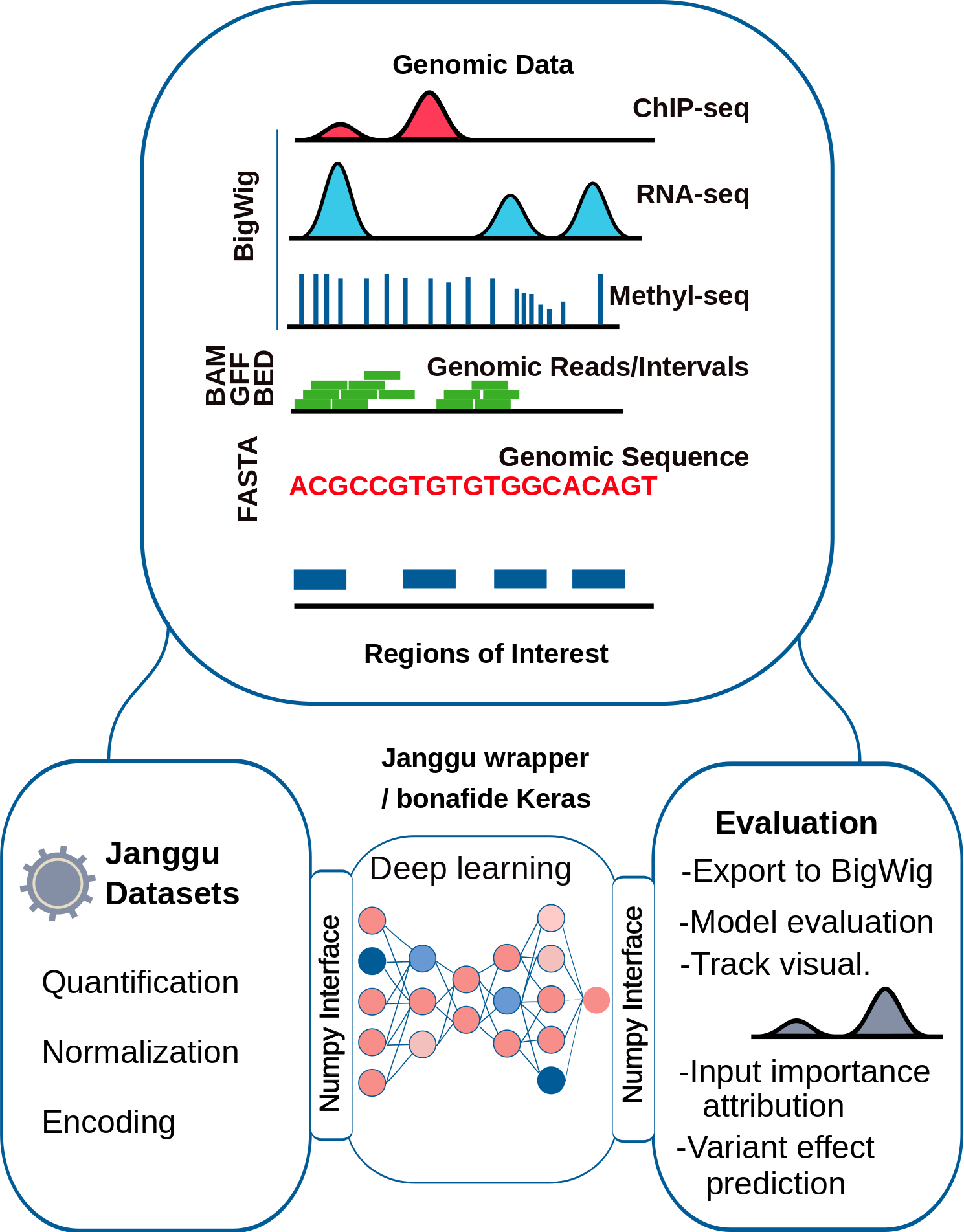
In particular, the package allows for easy access to typical Genomics data formats and out-of-the-box evaluation (for keras models specifically) so that you can concentrate on designing the neural network architecture for the purpose of quickly testing biological hypothesis. A comprehensive documentation is available here.
Hallmarks of Janggu:¶
- Janggu provides special Genomics datasets that allow you to access raw data in FASTA, BAM, BIGWIG, BED and GFF file format.
- Various normalization procedures are supported for dealing with of the genomics dataset, including ‘TPM’, ‘zscore’ or custom normalizers.
- Biological features can be represented in terms of higher-order sequence features, e.g. di-nucleotide based features.
- The dataset objects are directly consumable with neural networks for example implemented using keras or using scikit-learn (see src/examples in this repository).
- Numpy format output of a keras model can be converted to represent genomic coverage tracks, which allows exporting the predictions as BIGWIG files and visualization of genome browser-like plots.
- Genomic datasets can be stored in various ways, including as numpy array, sparse dataset or in hdf5 format.
- Caching of Genomic datasets avoids time consuming preprocessing steps and facilitates fast reloading.
- Janggu provides a wrapper for keras models with built-in logging functionality and automatized result evaluation.
- Janggu supports input feature importance attribution using the integrated gradients method and variant effect prediction assessment.
- Janggu provides a utilities such as keras layer for scanning both DNA strands for motif occurrences.
Getting started¶
Janggu makes it easy to access data from genomic file formats and utilize it for machine learning purposes.
dna = Bioseq.create_from_genome('dna', refgenome=<refgenome.fa>, roi=<roi.bed>)
labels = Cover.create_from_bed('labels', bedfiles=<labels.bed>, roi=<roi.bed>)
kerasmodel.fit(dna, labels)
A range of examples can be found in ‘./src/examples’ of this repository, which includes jupyter notebooks that illustrate Janggu’s functionality and how it can be used with popular deep learning frameworks, including keras, sklearn or pytorch.
Why the name Janggu?¶
Janggu is a Korean percussion instrument that looks like an hourglass.
Like the two ends of the instrument, the philosophy of the Janggu package is to help with the two ends of a deep learning application in genomics, namely data acquisition and evaluation.
Installation¶
A list of python dependencies is defined in setup.py. Additionally, bedtools is required for pybedtools which janggu depends on.
Janggu depends on tensorflow and keras. To install janggu with tensorflow version 1 and 2 use
# to install with tensorflow==1.14 and keras==2.2
pip install janggu[tf] # or janggu[tf_gpu]
# to install with tensorflow==2.2 and keras==2.4.3
pip install janggu[tf2] # or janggu[tf2_gpu]
Depending on the pip version (e.g. 20.2.2), some package dependencies may fail to be resolved accurately such that incompatible package versions are installed. If this is the case, you could try using pip install … –use-feature=2020-resolver or install the required package version manually.
Alternatively, you can install tensorflow and keras via the conda environment using
# tensorflow v1
conda install tensorflow==1.14 keras==2.2 # or tensorflow-gpu
# tensorflow v2
conda install tensorflow==2.2 keras==2.4.3 # or tensorflow-gpu
Further information regarding the installation of tensorflow can be found on the official tensorflow webpage
To verify that the installation works try to run the example contained in the janggu package as follows
git clone https://github.com/BIMSBbioinfo/janggu
cd janggu
python ./src/examples/classify_fasta.py single
A model is then trained to predict the class labels of two sets of toy sequencesby scanning the forward strand for sequence patterns and using an ordinary mono-nucleotide one-hot sequence encoding. The entire training process takes a few minutes on CPU backend. Eventually, some example prediction scores are shown for Oct4 and Mafk sequences. The accuracy should be around 85% and individual example prediction scores should tend to be higher for Oct4 than for Mafk.
You may also try to rerun the training by evaluating sequences features on both strands and using higher-order sequence encoding using i.e. the command-line arguments: dnaconv -order 2. Accuracies and prediction scores for the individual example sequences should improve compared to the previous example.